Unlocking Cancer Treatments: New Drug Design Targets 'Twin' Proteins
2024-10-01
Unlocking Cancer Treatments: New Drug Design Targets 'Twin' Proteins
In a groundbreaking study published in *Nature Chemical Biology*, Scripps Research scientists have revealed a revolutionary method for designing drugs that can effectively target elusive cancer-related proteins. While some proteins have obvious binding sites for drugs, others are notoriously difficult to access due to their structure. Enter the concept of "paralog hopping"—a strategy that could transform how we approach drug development.
Scientists have taken cues from the protein's paralog, or "twin", to uncover potential drug-binding sites. This innovative method allows researchers to identify ways to target proteins that previously seemed immune to drug intervention. Approximately half of the proteins in human cells have paralogs, many of which are involved in critical biological processes like cancer and autoimmune diseases. This means that the implications of their findings extend well beyond the initial study.
Senior author Benjamin Cravatt, a renowned chemist and biologist, highlighted the significance of targeting specific paralogs due to their differing functions. His team built upon findings that have arisen from the evolutionary phenomenon of gene duplication, which results in proteins that can perform overlapping roles in the body but have slight variations in their sequences.
The researchers pinpointed a unique approach by focusing on the amino acid cysteine, which has highly reactive properties that make it an exceptional target for drug binding. However, not all proteins possess accessible cysteines, prompting the need to creatively explore alternative binding sites on these twin proteins.
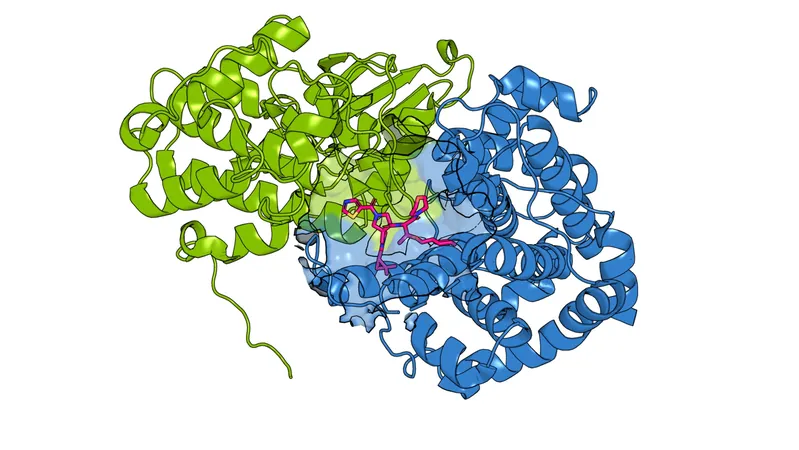
In their case study, Cravatt’s team focused on the CCNE1 and CCNE2 protein pair, known culprits in breast, ovarian, and lung cancers. Previous studies suggested that these proteins, while similar in structure, play distinct roles in tumor development. In an effort to selectively inactivate only one of the proteins, the team embarked on a quest to find appropriate binding sites.
After engineering a cysteine into CCNE1 to mimic a previously identified drug binding site on CCNE2, the researchers then screened a diverse library of chemical compounds. Their goal was to find drugs that could bind to CCNE1 without relying on the cysteine, ultimately revealing previously unknown binding pockets.
The results were astonishing, uncovering multiple compounds capable of binding to CCNE1 even in the absence of cysteine. Some compounds demonstrated unique properties, showing unexpected functions such as stabilizing the protein, which could lead to enhanced activity rather than deactivation. This emphasized the need for thorough screening of drug candidates via diverse methods that capture a wide range of functional molecules.
Looking ahead, Cravatt and Zhang's team is eager to apply their pioneering paralog-hopping method to other protein pairs that are implicated in cancer progression. Ongoing research will determine whether these newly discovered compounds can be utilized in therapeutic strategies against cancer and other diseases linked to CCNE1 and similar proteins.
This transformative approach not only paves the way for more targeted cancer therapies but also shines a spotlight on the potential for drug development inspired by nature's own complexities. With nearly half of human proteins being paralogs, the breakthroughs achieved by this study hold the promise of revolutionizing the field of drug design and cancer treatment.

 Brasil (PT)
Brasil (PT)
 Canada (EN)
Canada (EN)
 Chile (ES)
Chile (ES)
 España (ES)
España (ES)
 France (FR)
France (FR)
 Hong Kong (EN)
Hong Kong (EN)
 Italia (IT)
Italia (IT)
 日本 (JA)
日本 (JA)
 Magyarország (HU)
Magyarország (HU)
 Norge (NO)
Norge (NO)
 Polska (PL)
Polska (PL)
 Schweiz (DE)
Schweiz (DE)
 Singapore (EN)
Singapore (EN)
 Sverige (SV)
Sverige (SV)
 Suomi (FI)
Suomi (FI)
 Türkiye (TR)
Türkiye (TR)