Groundbreaking Study Links Gene Variations to Bicuspid Aortic Valve Disease!
2025-01-15
Author: John Tan
Groundbreaking Study Links Gene Variations to Bicuspid Aortic Valve Disease!
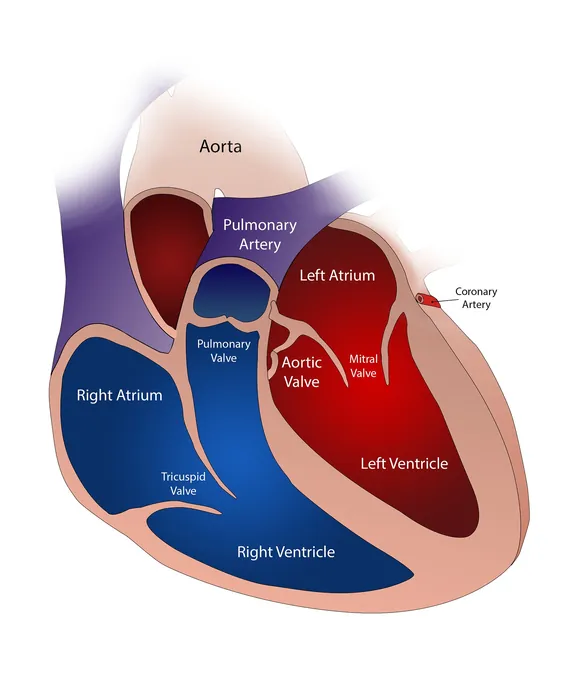
In a significant advancement for cardiac health, researchers from UTHealth Houston have uncovered that rare duplications and deletions within a specific chromosome region—22q11.2—are associated with nonsyndromic bicuspid aortic valve disease (BAV). This finding, recently published in the esteemed journal *Heart*, opens new avenues for understanding and potentially managing this common congenital heart defect.
Bicuspid aortic valve disease is characterized by the presence of only two leaflets in the aortic valve, rather than the normal three, impacting as much as 2% of the global population. While this condition is congenital and manifests from birth, it can often go undiagnosed until complications arise, leading to critical conditions such as thoracic aortic aneurysms, aortic stenosis, and other cardiovascular problems.
The research revealed that 7.4% of participants with early-onset BAV had rare genomic variations in the 22q11.2 region, which is crucial for regulating heart development. Co-first authors Sara Mansoorshahi and Catherina Tovar Pensa, both third-year medical students at McGovern Medical School, articulated their intent to bridge the gap in knowledge regarding these gene variations, potentially aiding in risk assessment and guiding clinical management for patients afflicted with this disease.
Previous studies have established a link between genetic duplications or deletions and congenital defects, but this research spotlights the 22q11.2 region specifically in relation to nonsyndromic BAV. The implications of these findings could be profound; if confirmed, they may influence how doctors evaluate and monitor patients for potential complications related to the condition.
Intriguingly, the 22q11.2 deletion syndrome, often linked with DiGeorge syndrome, stems from a deletion on chromosome 22 and can result in numerous defects—ranging from cardiac issues to learning difficulties and psychiatric conditions. Understanding the variants in this tip-top chromosome region is essential in establishing protocols for patients with BAV who may also exhibit early onset symptoms or secondary congenital malformations.
Employing whole genome microarray genotyping techniques, the researchers studied 272 BAV patients alongside their biological relatives, noting the copy number variations present. Key genes identified include TBX1, CRKL, HIC2, and MAPK1, all critical for proper vascular development. Notably, until this study, TBX1 had not been explicitly linked to BAV—a discovery that could reshape the diagnostic landscape for this heart defect.
This groundbreaking research not only enhances the understanding of nonsyndromic bicuspid aortic valve disease but also suggests that genetic testing for 22q11.2 copy number variants might soon become a standard practice for patients exhibiting these conditions. As the science advances, families affected by BAV might finally gain insight into potential risks and management strategies, leading to better outcomes and improved quality of life.
Stay tuned as further research unfolds to illuminate the genetics of congenital heart defects! This crucial information could transform lives and redefine medical approaches to these complex conditions.



 Brasil (PT)
Brasil (PT)
 Canada (EN)
Canada (EN)
 Chile (ES)
Chile (ES)
 Česko (CS)
Česko (CS)
 대한민국 (KO)
대한민국 (KO)
 España (ES)
España (ES)
 France (FR)
France (FR)
 Hong Kong (EN)
Hong Kong (EN)
 Italia (IT)
Italia (IT)
 日本 (JA)
日本 (JA)
 Magyarország (HU)
Magyarország (HU)
 Norge (NO)
Norge (NO)
 Polska (PL)
Polska (PL)
 Schweiz (DE)
Schweiz (DE)
 Singapore (EN)
Singapore (EN)
 Sverige (SV)
Sverige (SV)
 Suomi (FI)
Suomi (FI)
 Türkiye (TR)
Türkiye (TR)
 الإمارات العربية المتحدة (AR)
الإمارات العربية المتحدة (AR)