Unraveling GATA2 Deficiency and Its Deadly Link to Hemophagocytic Lymphohistiocytosis: A Comprehensive Case Review
2024-11-04
Author: Ming
Introduction
GATA2 deficiency is an autosomal dominant genetic disorder that increases predisposition to various infections, encompasses a range of clinical presentations, and can lead to a potentially life-threatening condition known as hemophagocytic lymphohistiocytosis (HLH). This systematic review meticulously evaluates documented cases of GATA2 deficiency associated with HLH, offering critical insights for healthcare professionals facing this challenging condition.
Methodology
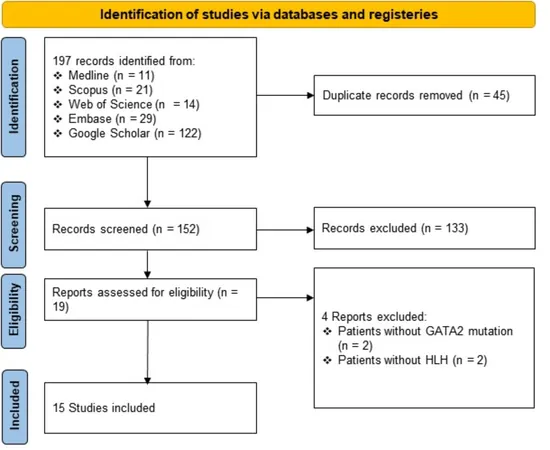
Following the PRISMA 2020 guidelines, our systematic literature review scrutinized case reports published from inception until June 14, 2024. We explored databases including Ovid MEDLINE ALL, Embase, Scopus, Web of Science, and Google Scholar, focusing on patients diagnosed with GATA2 deficiency who presented with HLH either concurrently or subsequently. Diverse study formats—ranging from original research papers to case series—were included, with no language restrictions.
Findings
Our review encompassed 15 studies from 2016 to 2024, involving 23 patients diagnosed with both GATA2 deficiency and HLH. The average age was 23.48 years, with a wide range spanning from 7 to 57 years. Notably, these patients exhibited a variety of genetic mutations and infection types, predominantly Mycobacterium avium, Mycobacterium kansasii, and several viral pathogens, including Epstein-Barr virus (EBV) and cytomegalovirus (CMV), all of which were implicated in precipitating HLH.
Hematopoietic stem cell transplantation (HSCT) was performed on 8 of the patients reviewed; 6 survived the procedure, though 2 did not. Alarmingly, the study identified a 39.13% overall mortality rate among GATA2 deficiency patients who experienced HLH.
HLH: A Severe and Deadly Condition
HLH, defined by Scott and Robb-Smith in 1939, remains a rare but devastating immunological disorder characterized by hyperactivation of immune cells, leading to an overwhelming inflammatory response that can damage multiple organ systems. Early detection and intervention are crucial, as HLH can progress rapidly to multiorgan failure.
Genetic predispositions are intricately linked to HLH, with over 100 genes identified to date, including those associated with cell-mediated immunity. GATA2 mutations stand out in this landscape as a significant contributor to both primary and secondary HLH cases.
The GATA2 Connection
GATA2, a transcription factor fundamental to hematopoiesis, is located on chromosome 3 and is critical for regulating blood cell development and immune function. More than 180 different mutations have been documented in GATA2, leading to varying clinical manifestations—including benign conditions like recurrent infections and severe ones such as myeloid malignancies.
Clinical challenges arise in managing patients with GATA2 deficiency due to their heightened vulnerability to infectious pathogens, which can serve as triggers for HLH. Certain infections commonly encountered include not just mycobacterial species, but also various bacterial, viral, and fungal pathogens.
Conclusion and Recommendations
This systematic review underscores the intricacies of GATA2 deficiency and its appropriate management, particularly concerning the risk of HLH. Key recommendations arise for clinicians, including:
1. Genetic Testing: Consider screening for GATA2 mutations in patients diagnosed with HLH.
2. Vigilance in Infection Management: Prioritize the early identification and treatment of infections to mitigate the risk of HLH development.
3. Monitoring Family Members: Given the hereditary nature of GATA2 mutations, screening first-degree relatives is vital to catch potential cases early.
Continued research into the intricate links between GATA2 deficiency and HLH is imperative for enhancing diagnostic accuracy and therapeutic strategies. Effective management remains critical to improving outcomes for patients faced with this challenging and multifaceted condition.
The study highlights the urgency of early intervention and careful monitoring post-transplant for patients with this genetic disorder.


 Brasil (PT)
Brasil (PT)
 Canada (EN)
Canada (EN)
 Chile (ES)
Chile (ES)
 España (ES)
España (ES)
 France (FR)
France (FR)
 Hong Kong (EN)
Hong Kong (EN)
 Italia (IT)
Italia (IT)
 日本 (JA)
日本 (JA)
 Magyarország (HU)
Magyarország (HU)
 Norge (NO)
Norge (NO)
 Polska (PL)
Polska (PL)
 Schweiz (DE)
Schweiz (DE)
 Singapore (EN)
Singapore (EN)
 Sverige (SV)
Sverige (SV)
 Suomi (FI)
Suomi (FI)
 Türkiye (TR)
Türkiye (TR)