Revolutionizing Material Science: A Breakthrough in Modeling Electric Responses with AI
2025-06-09
Author: Sarah
A Quantum Leap: Modeling Millions of Atoms
Researchers from the Harvard John A. Paulson School of Engineering and Applied Sciences (SEAS) have unveiled a groundbreaking machine learning framework capable of predicting how materials react to electric fields with astonishing quantum-level precision—scaling simulations up to a million atoms! This innovative leap far surpasses conventional quantum mechanical methods, which are limited to modeling only a few hundred atoms at a time.
Implications for Advanced Materials and Energy Technologies
This revolutionary approach will empower scientists and engineers to conduct highly accurate, large-scale simulations of how various materials respond to diverse external stimuli, paving the way for transformative advancements in materials design and cutting-edge energy technologies.
A Collaborative Effort in Research
The study was spearheaded by Stefano Falletta, a former postdoctoral researcher in the lab of Boris Kozinsky, the Gordon McKay Professor of Materials Science and Mechanical Engineering at SEAS, and Professor of Chemistry and Chemical Biology. Published in Nature Communications, this research addresses a significant challenge that has persisted for decades.
The Limitations of Traditional Methods
For over 30 years, density functional theory (DFT) has been the go-to method for simulating atomic and molecular behaviors. Although highly accurate, it is computationally demanding and restricted to smaller systems. Recent advancements in machine learning have made it possible to explore quantum behaviors at larger scales, but modeling responses to external stimuli remained a tough nut to crack.
Breaking New Ground with Machine Learning
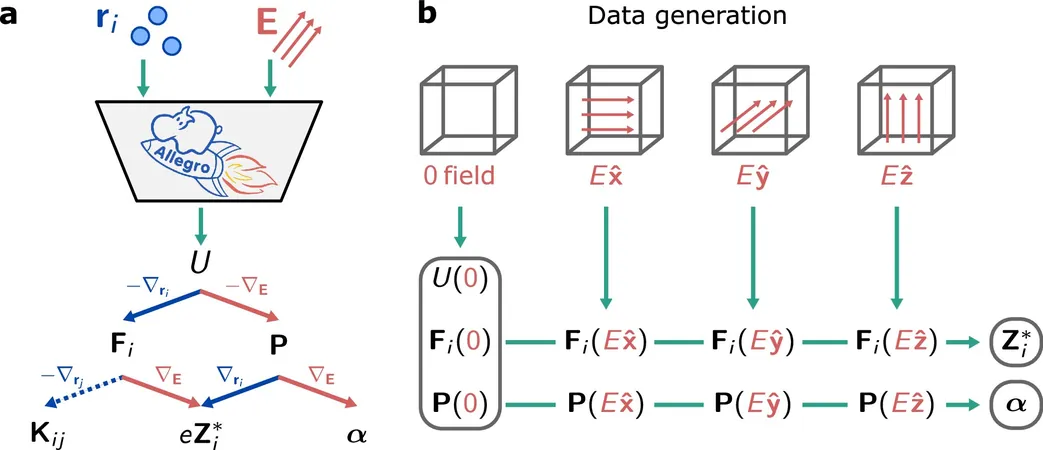
To tackle this, the researchers developed a novel machine learning approach that integrates disparate quantum behaviors—such as energy and polarization—into a singular, generalized potential energy function. This method utilizes density functional theory calculations for training and validation and incorporates the effects of external fields while ensuring adherence to fundamental physical principles.
Introducing Allegro-pol: The Future of Material Simulation
Named Allegro-pol, this new framework builds upon the previously established neural network architecture Allegro, which accurately simulates atomic energies and forces. Falletta aimed to extend Allegro's capabilities to include real-time molecular dynamics and to analyze the effects of external perturbations, like electric fields, on atom interactions.
Unlocking New Possibilities in Material Discovery
Understanding these interactions is crucial for discovering materials such as ferroelectrics and dielectrics, which have potential applications in non-volatile memory, capacitors, and energy storage devices. "Traditional physics-based methods are limited to modeling a few hundred atoms, but with our machine learning approach, we can stretch this to millions," explained Falletta.
Proven Success in Real-World Applications
The effectiveness of their method was demonstrated by simulating the infrared and electrical properties of silicon dioxide and examining the temperature-dependent ferroelectric switching in barium titanate.
A Bright Future for Materials Science
As Falletta continues his work at Radical AI to accelerate materials discovery, he believes that foundational models like the one outlined in this research can ignite a new era in machine learning-enabled materials science, leading to innovations that were once unimaginable.




 Brasil (PT)
Brasil (PT)
 Canada (EN)
Canada (EN)
 Chile (ES)
Chile (ES)
 Česko (CS)
Česko (CS)
 대한민국 (KO)
대한민국 (KO)
 España (ES)
España (ES)
 France (FR)
France (FR)
 Hong Kong (EN)
Hong Kong (EN)
 Italia (IT)
Italia (IT)
 日本 (JA)
日本 (JA)
 Magyarország (HU)
Magyarország (HU)
 Norge (NO)
Norge (NO)
 Polska (PL)
Polska (PL)
 Schweiz (DE)
Schweiz (DE)
 Singapore (EN)
Singapore (EN)
 Sverige (SV)
Sverige (SV)
 Suomi (FI)
Suomi (FI)
 Türkiye (TR)
Türkiye (TR)
 الإمارات العربية المتحدة (AR)
الإمارات العربية المتحدة (AR)